Research
Research
Soft Matter Simulations in the Wilson Group
Contact Details
Prof. Mark Wilson
Department of Chemistry
University Science Laboratories
South Road
Durham
DH1 3LE
United Kingdom
Tel: +44 (0) 191 334 2144
Fax: +44 (0) 191 384 4737
Email: [email protected]
| Liquid Crystals | Home |
| Amphiphiles | Mark Wilson |
| Macromolecules | Publications |
| Coarse-graining | Talks |
| Chirality | Chemistry Dept |
| Parallel Simulations | Durham University |
Research Summary
We use classical molecular dynamics and Monte Carlo simulation methods to study problems in soft matter chemistry, concentrating on materials that are ordered on the nanoscale. Our simulations look at:
- structure and dynamics in chromonic liquid crystals,
- structure and dynamics in thermotropic nematics (including biaxial nematics),
- models to understand dynamic allostery in proteins,
- models to understand the structure of membranes,
- molecular interactions between amphiphilic molecules in water (including lipids and amphiphilic polymer systems),
- chirality transfer in liquid crystals,
- the relationship between molecular structure and bulk structure in polyphilic polymers and dendrimers,
- methods for “coarse-graining”, of complex chemical systems,
- dynamics in soft solids,
- developing methods for parallel simulation and testing new methods for molecular simulation (including metadynamics and replica exchange).
This page contains a brief summary of some of our recent research projects, with links to more details. See also our publications page to find out more about the research work we do.
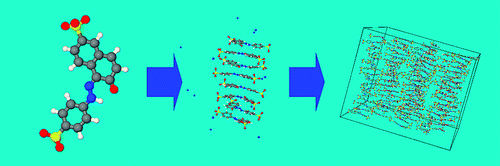
Simulation of chromonic molecules and chromonic phases
{kind=link}
We have recently1 carried out the first detailed study of the structure and dynamics of a chromonic liquid crystal (edicol – the food dye sunset yellow). The simulations show spontaneous self-assembly in water at atomistic detail, and can be used to determine the free energy of association in solution. Sunset yellow is seen to form single molecule stacks with a preference for head-to-tail ordering, which at higher concentrations form liquid crystal phases in water (as shown in the picture above). Our work shows that simulation can be used to understand, predict and control molecular self-assembly with a view to the future use of chromonic materials for applications in self-assembled molecular electronics.
Read More (2010 JACS paper).
Models to understand dynamic allostery in proteins
{kind=link}
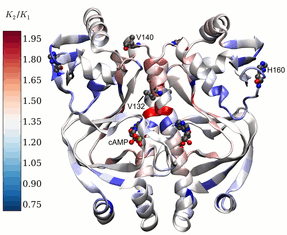
Allostery is a process by which a molecule binding to one site of a protein alters the activity of the protein at another site. We have developed new models to help understand how protein dynamics can influence allostery. In cases where the protein undergoes no conformational change on binding a small molecule, we demonstrate that that allostery arises as a natural consequence of changes in global low-frequency protein fluctuations on ligand binding.
Read More (PLoS Biol, 2013 paper).
Study of the structure and dynamics of molecules in biological membranes
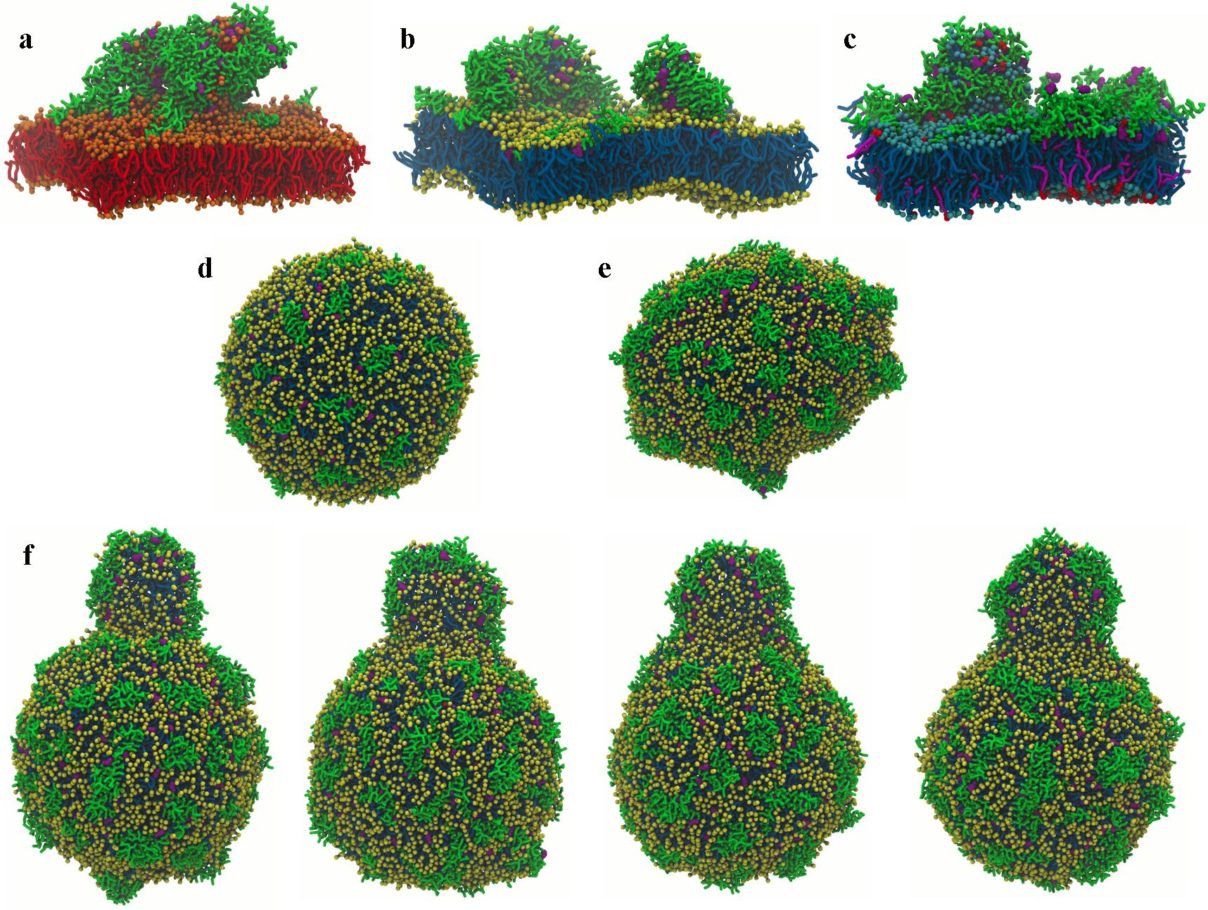
With our collaborators at UEA and the SOFI CDT , we have been developing models to understand the structure and dynamics of molecules within lipid membranes. This is of fundamental importance in trying to understand many processes that occur within cell membranes.
We are using a combination of atomistic simulations that provide an “atom-level” picture within the membrane, and coarse-grained sinulations that, at a slightly less detailed level, provide information on processes that occur on long timescales (such as the formation of lipid rafts and membrane pores).
We are also using these models to try and understand how antimicrobial ageents, such as antimicrobial peptides and peptoids, can lead to membrane lysis.
{kind=link}
Simulation of thermotropic liquid crystal molecules
Biaxial nematics
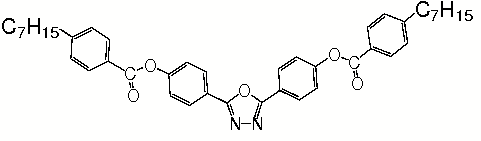
ODBP-Ph-C7. The first “true” low molecular weight biaxial nematic.

We have2 used atomistic simulation to study the structure of “the first” low molecular weight biaxial liquid crystal, ODBP-Ph-C7. The sequence shows a sequence of time frames showing the slow growth of a biaxial phase from a uniaxial starting point. As the phase grows it develops ferroelectric domains, which are shown colour-coded. Molecules which are blue and red have transverse dipoles pointing in opposite directions.
Read More (2006 PRL on biaxial nematics)
{kind=link}
Studying dynamics

We have also (with our collaborators at UEA) been developing techniques to understand dynamics in liquid crystals and to use simulation as a tool to help interpret experimental measurements of dynamical processes.3,4
Read More (PRL 2009).Read More (Chem Eur. J. 2010).
We have developed new methods for calculating material properties in liquid crystal phases from atomistic simulations. For example, the rotational viscosity determines how quickly a nematic liquid crystal can reorientate in a liquid crystal display; and can be determined from our simulations by monitoring the liquid crystal director in the course of a molecular dynamics simulation.5 To carry out this work, we need accurate representations of intramolecular interactions within a molecule and intermolecular interactions between molecules. This is made possible by a new force field for use in materials chemistry, which we have been developing.6
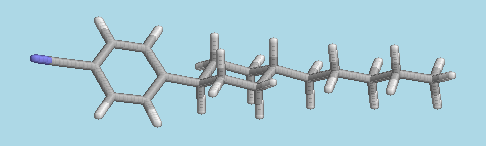 A snapshot from a molecular dynamics simulation showing the liquid crystal molecule PCH5 in a nematic phase.
A snapshot from a molecular dynamics simulation showing the liquid crystal molecule PCH5 in a nematic phase.
Coarse-grained models for the simulation of polymers, dendrimers, liquid crystals and lipids
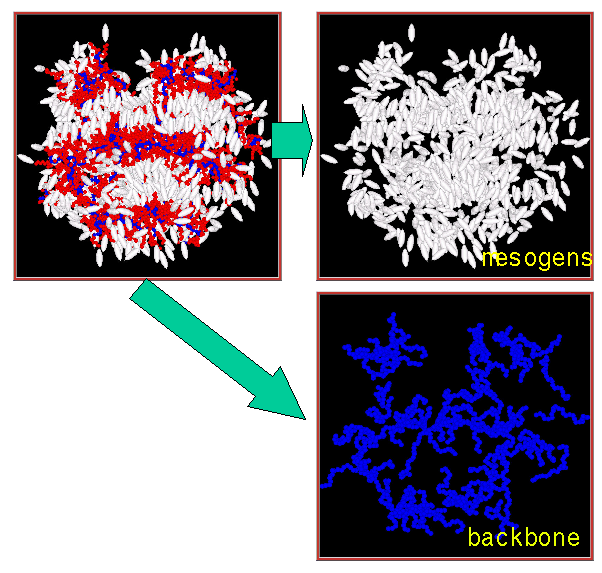
We have been developing coarse-grained simulation methods that allow us to understand structure and organisation in a range of molecular materials. We have recently studied liquid crystalline polymers and dendrimers and are working on “molecular engineering” tools to design molecules to have the desired organisation at the nanoscale. This research is being extended to lipid bilayers and vesicles. An important part of our work has involved developing efficient simulation methods that make use of parallel computers, so we are able to simulate extremely large systems composed of many molecules and take advantage of today’s supercomputers.
The figure (left) shows microphase separation in a liquid crystalline side chain polymer to give polymer-rich and liquid crystal-rich regions. Blue: polymer backbone (methylsiloxane), white: liquid crystal group, red: flexible spacer groups (alkyl chain).

The pictures (left) show the structure of a series of mesophases formed by a multiblock copolymer as studied by coarse-grained modelling.7 The phases are nematic (top left), lamellar (top right and middle right for different lengths of rods), gyroid (middle left), micellar (bottom). The bottom left structure show elongated micelles formed from polymer rods.

The second picture shows the formation of a polymer nanowire within a simulation.
Such wires may well have future applications in the area of nanoelectronics. Read More.
Calculations of helical twisting power – chirality mediated by intermolecular forces
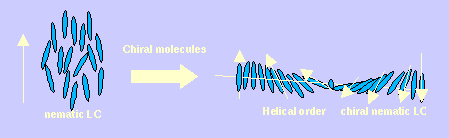
When a nematic liquid crystal phase is doped with a small amount of a chiral molecule, the whole phase becomes chiral, and the liquid crystal director (which describes the preferred direction of alignment for the molecules) is twisted through space to form a helix. Some molecules are better than others at inducing this twist, and the effectiveness of an individual molecule is measured by its Helical Twisting Power (HTP). There is considerable industrial interest in making new molecules with very large HTPs, to use as chiral dopants for chiral polymer films. However, it is difficult to relate chemical structure to HTP.
We developed new methods for predicting HTP prior to synthesis.
Our work suggests that each separate molecular conformation has a different HTP, and we can show how the overall HTP value changes with increases in temperature (as higher energy conformations with different HTPs start to become populated). This mechanism explains the temperature-induced reversal in HTP that occurs with some materials. It also explains the fact that some molecules can have different HTPs in different solvents. The latter is caused by some conformations being preferentially selected in certain solvents.
Recently, we have applied out methods to “banana molecules” and the results help explain the remarkable observation that bananas can behave as if they are chiral, even though the molecules themselves are achiral.Read More.
Dynamics in soft solids
{kind=link}
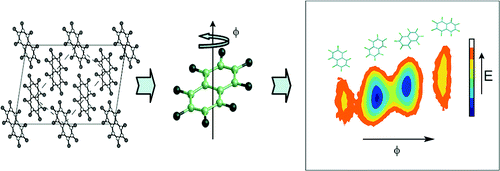
We have been using atomistic simulation to study dynamics in molecular solids. Our recent work with octafluoronaphthalene and urea inclusion compounds has indicated that its is now possible to look at dynamical processes over tens of ns, and study correlated motion in solids. This provides the possiblity of using simulation to complement and help interpret experimental solid state NMR data, and raises intriguing questions as to whether we can use simulation to help engineer tractable molecular machines for future applications.
Read More (2010 JACS paper).
Read More (2011 JPC paper).
Simulation of amphiphilic molecules at a water-air interface
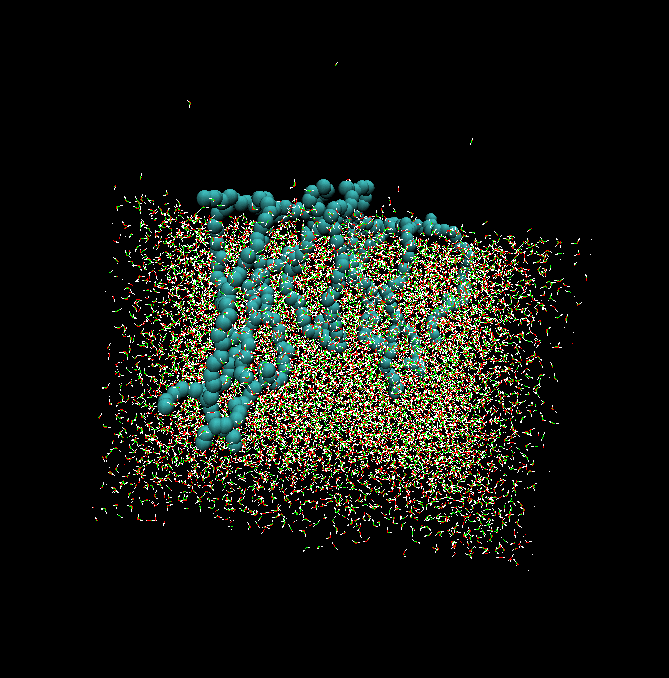
{kind=link}
We are interested in the properties of amphiphilic molecules and have used simulation to understand the behaviour of amphiphilic polymers at a water/air interface.8 The simulations can be used to interpret the results of complementary neutron diffraction studies by calculating neutron reflectivity curves to match with experiment.
The snapshot (left )is from a simulation study of an amphiphilic polymer at a water-air interface (see cartoon below). At higher surface concentrations the PEO chains are forced down into the aqueous subphase to form a polymer brush.
Similar modelling techniques are being used to study lipid – amino acid interactions. Our recent atomistic work in this area seeks to understand how these interactions (specifically the balance of hydrogen bonding and cation-Pi interactions) can be perturbed by small chemical modifications of the amino acid structure. Such studies seek to aid our understanding of binding between proteins and cell membranes.
Parallel Simulation Techniques
A key part of our work involves the design and implementation of parallel simulation techniques. These techniques allow computational problems to be tackled by an array of powerful processors working together. We have recently developed new computer codes which allow molecular dynamics and Monte Carlo simulations to be carried out in parallel. These techniques are readily translated to other parallel systems, paving the way for a new generation of large-scale simulations of chemical systems. Our current work in this area involves in the design of new methods for the large-scale simulation of polymer systems and the development of parallel tempering methods. More information about our programs is available from our software page.
Read More.
Key References
- Chami F. and Wilson M. R. Molecular Order in a Chromonic Liquid Crystal: A Molecular Simulation Study of the Anionic Azo Dye Sunset Yellow J. Am. Chem. Soc., 2010, 132, 7794.
- Pelaez J. and Wilson M. R. Atomistic Simulations of a Thermotropic Biaxial Liquid Crystal. Phys. Rev. Lett., 2006, 97., 267801.
- Oganesyan, V. S., Kuprusevicius, E., Gopee, H., Cammidge, A. N., Wilson, M. R., Simulation of EPR spectra directly from molecular dynamics trajectories of a liquid crystal with doped paramagnetic spin probe. Phys. Rev. Lett., 2009, 102, 013005(1-4)
- Oganesyan, V. S., Kuprusevicius, E., Gopee, H., Cammidge, A. N., Wilson, M. R., Prediction of EPR Spectra of Liquid Crystals with Doped Spin Probes from Fully Atomistic Molecular Dynamics Simulations: Exploring Molecular Order and Dynamics at the Phase Transition. Chem. Eur. J., 2010, 16, 11558
- Cheung D. L., Clark S. J, Wilson M. R. Calculation of the rotational viscosity of a nematic liquid crystal. Chem. Phys. Lett., 2002, 356, 140.
- Cheung D. L., Clark S. J., Wilson M. R. Parametrization and validation of a force field for liquid-crystal forming molecules . Phys. Rev. E, 2002, 65, 051709.
- Lintuvuori, J. S., Wilson M. R. A coarse-grained simulation study of mesophase formation in a series of rod-coil multiblock copolymers. , Phys. Chem. Chem. Phys., 2009, 11, 2116 – 2125.
DOI:10.1039/b818616b. - Anderson P. M. and Wilson M. R. Molecular Dynamics Simulations of an Amphiphilic Graft Copolymer at a Water/Air Interface., 2004, J. Chem. Phys, 121, 8503.
- Wilson M. R., Ilnytskyi J. M., Stimson L. M., Computer simulations of a liquid crystalline dendrimer in liquid crystalline solvents. J. Chem. Phys., 2003, 119, 3509.
- Ilnytskyi J, Wilson M. R.. A domain decomposition molecular dynamics program for the simulation of flexible molecules with an arbitrary topology of Lennard-Jones and/or Gay-Berne sites. Comput. Phys. Comm., 2001, 134, 23.
- Earl D. J., M., Wilson M. R. Predictions of molecular chirality and helical twisting powers: A theoretical study. J. Chem. Phys., 2003, 119, 10280.
- Ilott A. J., Palucha S., Batsanov A. S., Wilson M. R. and Hodgkinson P. Elucidation of Structure and Dynamics in Solid Octafluoronaphthalene from Combined NMR, Diffraction, And Molecular Dynamics Studies. J. Am. Chem. Soc., 2010, 132, 5179.
- Ilott A. J., Palucha S., Batsanov A. S., Harris, K. D. M., Hodgkinson P., Wilson M. R, Structural Properties of Carboxylic Acid Dimers Confined within the Urea Tunnel Structure: An MD Simulation Study. J. Phys. Chem., 2011, 115, 2791.